Maria Cláudia Nogueira Zerbini
Introdução: Com base nos resultados de estudos prévios, o Grupo de estudo dos tumores renais do Sociedade Internacional de Oncologia Pediátrica (SIOP-RTSG) desenvolveu um novo protocolo de estudo para os tumores renais pediátricos: o UMBRELLA SIOP-RTSG 2016 (o protocolo UMBRELLA). Esse protocolo é o adotado pela Sociedade Brasileira de Oncologia Pediátrica (SOBOPE), como protocolo SIOP-RTSG-GBTR 2016. Recomenda-se a leitura das referencias citadas neste capítulo, com especial atenção para aquelas destacadas em negrito. Mais do que nunca, o sucesso terapêutico no tratamento do TW depende do cuidado do patologista em seguir detalhadamente cada item do processamento e diagnóstico desses espécimes. Idealmente, após o diagnóstico pelo patologista local, o material (lâminas e 1 bloco de parafina) deve ser encaminhado sempre para o GBTR/SOBOPE o mais breve possível, para revisão patológica central junto a cópia do laudo (ver referência).
Código de topografia
C64 Neoplasia maligna do rim, exceto pelve renal e carcinoma de células renais
I.Identificação
II. Informes clínicos
- História familiar de tumores (renais) (especificar) __________
- Paciente portador de síndrome clínica, malformações/outras neoplasias ≈
- Beckwith-Wiedemann
- da Nefroblastematose familiar de Perlman
- de Denys-Drash
- Trissomia 18
- Neurofibromatose
- Síndrome de Bloom
- Síndrome EAGR
- Outra: Especificar__________________
Biópsia prévia a cirurgia
- Não
- Sim (especificar tipo)
- PAAF___ □ Agulha grossa ___ □ Cunha____
- Desconhecido
Quimioterapia pré-operatória
- Sim (especificar) __________(drogas) __________(número de ciclos)
- Não
- Desconhecido
Metástases a distância
- Não
- Sim (especificar) __________
- Desconhecido
Procedimento
- Nefrectomia parcial
- Nefrectomia radical
- Nefrectomia parcial bilateral
- Outro (especificar) __________
Lateralidade
- Direito
- Esquerdo
- Não especificado
Outras informações relevantes (especificar) __________
III. Exame macroscópico/representação histológica
Considerações iniciais importantes no manuseio do espécime de nefrectomia por tumor renal na infância:
- O material da nefrectomia deve ser remetido intacto ao patologista pelo cirurgião; em hipótese nenhuma deve ser violado. O patologista deve se eximir de fornecer o estadiamento anatomopatológico caso isso ocorra, pois o planejamento terapêutico, no que se refere à radioterapia, baseia-se principalmente no estadiamento anatomopatológico local
- O espécime deve ser pesado, medido e fotografado
- Áreas suspeitas de ruptura ou invasão devem ser identificadas e especificadas na requisição pelo cirurgião
- A cápsula do rim/tumor nunca deve ser retirada
- Os linfonodos (LN) peri-hilares e perirrenais devem ser pesquisados e representados para exame microscópico de forma identificada
- A superfície de todo o espécime deve ser pintada e seca antes de abrir a peça. Lembrar que o estadiamento local é crucial para a definição do prognóstico e decisão terapêutica e depende essencialmente desses cuidados iniciais
- O espécime deve ser aberto por uma incisão longitudinal para revelar o tumor e sua relação com o rim, cápsula renal e seio renal
- A superfície de corte deve ser fotografada, descrevendo-se o aspecto macroscópico
- O tumor deve ser medido. Nos espécimes pós-quimioterapia, a extensão da necrose deve ser estimada.
- O espécime deve ser fixado em formol a 4% tamponado por no máximo 24 a 48 horas. Fatias paralelas adicionais podem ser realizadas no espécime para melhor fixação.
- A coleta do material para estudo genético deve ser feita pelo patologista, sem prejuízo ao estadiamento anatomopatológico.
III.1 Dimensões do espécime (rim)
Diâmetros ___ x ___ x ___
Peso ____ g
III.2 Localização do tumor nos rins (selecionar todas as opções que se aplicam ao caso)
- Polo superior
- Localização mediana
- Polo inferior
- Outra (especificar) __________
- Não especificada
III.3 Focalidade
- Unifocal
- Multifocal
- Número de tumores no espécime __________
- Não avaliável
III.4 Dimensões do tumor
- Maior diâmetro____ cm
- Dimensões adicionais ___ x ___ cm
- Não podem ser determinadas
Para espécimes com tumores múltiplos, especificar o maior diâmetro de cada tumor.
Exemplo:
Maior diâmetro do tumor 2 ____ cm
Maior diâmetro do tumor 3 ____ cm (e assim por diante)
III.5 Extensão macroscópica do tumor
Fáscia de Gerota
- Intacta
- Rota
- Indeterminada
- Não aferida
Seio renal* (selecionar todas as opções que se aplicam ao caso)
- Não envolvido pelo tumor
- Tecidos moles minimamente envolvidos pelo tumor
- Tecidos moles do seio extensamente envolvido
- Envolvimento das estruturas vasculares do seio renal (vasos linfáticos e/ou sanguíneos)
*SR: o seio renal é aquela área no hilo renal que contem a pelve renal, os vasos hilares e a gordura; nessa região a córtex renal não é revestida por cápsula. O SR estende-se da região hilar para o interior do rim.
Veia renal
- Invasão presente
- Invasão não identificada
- Indeterminada
- Não avaliável
Envolvimento de órgão adjacente
- Extensão do tumor a órgão adjacente presente (especificar o órgão) __________
- Extensão do tumor a órgão adjacente não identificada
- Indeterminada
- Não avaliável
III.6 Representação histológica
- Mapear a fatia mais representativa da neoplasia e suas relações com o parênquima renal, seio renal e cápsula – representar cortes designados de acordo com o desenho/identificação com documentação fotográfica que deve constar no laudo e ser detalhadamente descrita no relatório AP; a foto com as designações deve, sempre que possível, integrar o laudo. (Figura 1)
- A cápsula da nefrectomia nunca deve ser retirada, pois a invasão do tumor e a transposição capsular são critérios para o estadiamento; além disso, os restos nefrogênicos são frequentemente sub capsulares em localização
- Representar designadamente de maneira exaustiva as áreas de possível extensão extra renal ou áreas suspeitas de margens comprometidas, assim como qualquer área “diferente” ao exame macroscópico.
- Interface rim/tumor
- Interface tumor/seio renal.
- Nos tumores multicêntricos, representar cada nódulo.
- Rim não neoplásico: pelo menos um fragmento ou qualquer área suspeita de resto nefrogênico (RN) (em geral áreas mais pálidas do que o parênquima usual; mais de 30-40% dos espécimes de nefrectomia por TW contem RN))
- Margens: gordura hilar (margem do seio renal), artéria renal, veia renal, ureter
- Glândula suprarrenal – se presente
- Linfonodos – todos, designados.
- Gordura – se for enviada separadamente incluir na totalidade
- Outros (especificar) __________
- A coleta de material para estudo genético deve ser realizada sempre que possível, segundo orientação estabelecida pelo protocolo do GBTR/SIOP-RTSG, 2016 (ver referencia).
IV. Exame microscópico
IV.1. Critérios histológicos para subtipar o tumor de Wilms nos pacientes tratados com QT pré-operatória segundo protocolo da SIOP-RTSG, 2016 (Quadro 1)
Quadro 1: Critérios histológicos para a subclassificação histológica do TW nos pacientes submetidos a Qt prévia (SIOP-RTSG, 2016)
| Tipo histológico do tumor | Alterações induzidas pela Qt* (%) | Características histológicas (% de tumor viável) | ||
| Blastema | Epitelio | Estroma* | ||
| Completamente necrótico* | 100 | 0 | 0 | 0 |
| Regressivo*** | >66 | 0-100 | 0-100 | 0-100 |
| Misto | <66 | 0-65 | 0-65 | 0-65 |
| Misto | <66 | 11-65 | 0-89 | 0-89 |
| Epitelial | <66 | 0-33 | 66-100 | 0-33 |
| Estromal | <66 | 0-33 | 0-33 | 66-100 |
| Blastematoso | <66 | 66-100 | 0-33 | 0-33 |
*O tipo estromal em geral mostra apenas alterações induzidas pela QT mínimas ou moderadas, pelo fato do tecido estromal ser resistente a Qt.
** Tumor composto somente de alterações regressivas e/ou necróticas causadas pela Qt. A presença de túbulos maturos, estroma e pequenos grupos de blastema é aceita, podendo representar restos nefrogênicas, e não deve ser interpretada como tumor viável. A estimativa de necrose/alterações regressivas é feita macro e microscopicamente.
***Alterações regressivas compreendem uma mistura de necrose, fibrose, estroma fibromixóide contendo macrófagos com conteúdo lipídico (foam cells) ou de hemosiderina.
IV.2 Estratificação em grupos de risco com finalidade terapêutica
Atualmente, a Sociedade Brasileira de Oncologia Pediátrica (Sobope), para tratamento de tumor de Wilms (TW), segue o protocolo SIOP 2016 (Sociedade Internacional de Oncologia Pediátrica), que preconiza o tratamento quimioterápico pré-operatório sub classificando os tumores em grupos de risco.
As bases para a estratificação terapêutica levam em consideração a idade do paciente, o tipo do umos, subtipo histológico (grupo de risco histológico) e o estadiamento; nos nefroblastomas associam-se a esses parâmetros a presença de anaplasia, a proporção do componente blastematoso e de necrose/alterações regressivas (Quadro 2). Esse sistema de estratificação só é possível se for examinadao um número adequado de preparados histológicos, segundo i protocolo do estudo UMBRELLA.
Quadro 2 Estratificação prognóstica dos pacientes com tumores renais pediátricos (SIOP-RTSG, 2016).
| A. Para casos previamente tratados com Qt |
| I. Baixo risco |
|
Nefroma mesoblástico |
| Nefroblastoma cístico parcialmente diferenciado (tratamento cirúrgico apenas) |
| Nefroblastoma completamente necrótico |
| II. Risco intermediário |
| Nefroblastoma estromal |
| Nefroblastoma misto |
| Nefroblastoma com anaplasia focal |
| III. Alto risco |
|
Nefroblastoma blastematoso |
| Nefroblastoma com anaplasia difusa |
| Sarcoma de células claras |
| Tumor rabdoide renal
|
| B. Para nefrectomias primárias (sem Qt prévia) |
| I. Baixo risco |
|
Nefroma mesoblástico |
| Nefroblastoma cístico parcialmente diferenciado |
| II. Risco intermediário |
| Nefroblastoma não anaplásico e suas variantes |
| Nefroblastoma com anaplasia focal |
| III. Alto risco |
| Nefroblastoma com anaplasia difusa (independente de qualquer outra característica) |
| Sarcoma de células claras |
| Tumor rabdoide renal |
IV.3 Especificar outros tumores mais raros (ver VI.2)
IV.4. Restos nefrogênicos (RN) (selecionar todas as alternativas que se aplicam ao caso) (no caso de TW)
- RN não identificados
- RN presentes
- RN intralobares
- RN perilobares
- Difusos hiperplásicos
- Multifocais
- Focais
- RN inclassificáveis
- Não avaliáveis
Nota: A ausência de cápsula é um importante critério no caso de dúvida entre RN e TW.
IV.5. Margens
- Envolvimento não identificado das margens
- Menor distância entre o tumor e a margem _____ cm
- Especificar as margens ____________
- Margem envolvida pelo tumor
- Fáscia de Gerota
- Margem medial do seio renal
- Veia renal*
- Artéria renal
- Veia cava inferior*
- Ureter
- Outra (especificar) __________
* Notas: Cuidado deve ser tomado ao se avaliar a margem venosa quando existe trombo no interior do vaso, pois pode haver retração da veia após a exérese do tumor pelo cirurgião com protrusão do trombo, que pode ser erroneamente interpretado como margem da parede venosa comprometida pelo tumor. Sempre discutir com o cirurgião.
Importante: A presença de tumor não viável ou alterações regressivas somente na margem de ressecção não deve ser considerada margem comprometida (estádio III).
IV.6. Linfonodos (LN)*
- Metástases para linfonodos regionais não identificadas
- Metástases para linfonodos regionais presentes (especificar local) __________
- Linfonodos não enviados e não encontrados junto à peça
Número de linfonodos examinados (especificar) __________
Não podem ser determinados (explicar) __________
Número de linfonodos envolvidos (especificar) __________
- Metástases a distância
- Ausentes
- Presentes (especificar) __________
- Desconhecida
*A presença de alterações induzidas pela Qt (com ou sem tumor viável) em um linfonodo deve ser interpretada como metástase (estádio III) Metástase não viável é considerada quando macrófagos xantomatosos e/ou outras alterações induzidas pela QT substituem a arquitetura normal do LN.
V. Diagnóstico / tipo histológico
Grupo de risco __________
Estádio anatomopatológico __________ Justificativa __________
Comentários _______________________________________________________________
Foto (opcional): designação dos fragmentos representados para exame histológico (enfaticamente recomendada)
Patologista responsável __________________________CRM __________
VI. Informações adicionais
Os tumores renais constituem 7% dos tumores malignos pediátricos, sendo diagnosticados nos Estados Unidos cerca de 500 novos casos ao ano. Entre eles, os nefroblastomas (tumor de Wilms) correspondem a 85% dos casos, sendo os demais tipos histológicos, como o nefroma mesoblástico (5%), sarcoma de células claras (4%) e o tumor rabdoide (2%), bem menos frequentes.
O nefroblastoma (tumor de Wilms) acomete preferencialmente indivíduos entre 1 e 5 anos de idade. O progresso da poliquimioterapia e a formação de grupos cooperativos nos últimos 40 anos, como o vinculado à International Society of Pediatric Oncology (SIOP) e o National Wilms Tumor Study Group (NWTS), mais recentemente continuado pelo Children’s Oncology Group (COG) nos Estados Unidos, e o Grupo Brasileiro para o Tratamento do Tumor de Wilms (GBTTW), posteriormente GBTR, colaboraram de forma importante para que esse tipo de neoplasia se tornasse um exemplo de sucesso terapêutico. Dessa forma, a sobrevida em 5 anos, que se restringia a 8% no início do século XX, atingiu os 50% em 1960 e 90% no início do século XXI.
A formação dos grupos cooperativos para o estudo dos tumores renais pediátricos visa basicamente a:
- Centralizar casos de doenças raras contribuindo para um melhor entendimento dessas entidades.
- Diagnosticar adequadamente e classificar os tumores.
- Identificar aspectos que se relacionem ao comportamento biológico (marcadores de agressividade).
- Avaliar as resposta à QT e as margens de ressecção cirúrgicas.
- Estadiar a neoplasia da forma mais adequada à biologia da doença.
- Identificar aspectos epidemiológicos dos tumores renais pediátricos.
- Avaliar as complicações tardias do tratamento nas crianças sobreviventes ao tratamento para o tumor de Wilms.
VI.1 Nefroblastoma – conceitos importantes
VI.1.1 Anaplasia
No TW, a identificação de anaplasia, segundo os critérios citológicos propostos por Beckwith e Palmer, constitui fator prognóstico significativo, categorizando o tumor como de histologia desfavorável. O diagnóstico de anaplasia requer:
- Hipercromasia nuclear.
- Cariomegalia significativa, com núcleos pelo menos três vezes maiores que os das células tumorais adjacentes em pelo menos dois diâmetros.
- Figuras de mitoses multipolares e inequívocas.
A anaplasia correlaciona-se à resposta à quimioterapia, devendo ser classificada como focal ou difusa, utilizando-se os critérios propostos por Faria et al. (1996).
VI.1.1.1 Anaplasia focal
- Confinada a um ou mais focos bem delimitados dentro do tumor primário.
- Os focos de anaplasia não devem exceder 15mm.
- Ausente além da cápsula renal ou em sítios extrarrenais.
- Se mais de um foco estiver presente no tumor primário e for rodeado por tumor não anaplásico, sem atipia de fundo (nuclear unrest).
- Em pacientes previamente tratados, é confinada a um discreto foco rodeada por tumor necrótico.
VI.1.1.2 Anaplasia difusa
- Não localizada e/ou além da cápsula renal.
- Acentuada atipia em outras áreas do tumor (nuclear unrest).
- Não demarcada do tumor não anaplásico.
- Presente em sítios extra renais, seio renal, êmbolos ou metástases.
- Presente em uma biópsia randomizada.
VI.1.2 Restos nefrogênicos (RN)
Os restos nefrogênicos (RN) correspondem a áreas de tecido renal embrionário anormalmente persistentes. Eles podem apresentar diferentes evoluções: permanecer quiescentes, involuir, esclerosar, sofrer hiperplasia ou dar origem a neoplasias. Os RN estão presentes em cerca de 30% dos espécimes ressecados com TW. A presença de RN correlaciona-se com risco aumentado de tumor sincrônico ou metacrônico no rim contralateral.
Os RN podem ser classificados em:
- Perilobares – situados na periferia do lobo renal, apresentando margens bem definidas, compostos por blastema ou túbulos, com estroma escasso ou esclerótico, sendo geralmente múltiplos.
- Intralobares – situados ao acaso na cortical ou medular, com margens indistintas, dispersos entre os néfrons, compostos de blastema e túbulos, porém com mais estroma do que os perilobares, geralmente únicos e limitados à margem do tumor.
- Nefroblastematose – presença difusa ou multifocal de RN ou de seus derivados – recomendada QT prolongada.
- A multicentricidade (tumores múltiplos) observada no espécime da nefrectomia deve ser analisada nesse contexto. Deve-se, portanto, amostrar o rim normal (não tumoral), visando a identificar RN.
A Tabela 3 resume as principais características dos restos nefrogênicos perilobares e intralobares.
Quadro 3. Características dos RN perilobares e intralobares
| Característica | RN perilobar | RN intralobar |
| Local no lobo renal | Periferia | Córtex, medula, seio renal |
| Margem | Bem demarcada | Pouco demarcada |
| Relação com néfrons | Néfrons ausentes nos RN | RN dispersos entre os néfrons |
| Composição | Blastema ou túbulos; estroma escasso ou esclerótico | Túbulos, blastema, cistos; predominância de estroma em geral |
| Número | Geralmente numerosos | Frequentemente único |
| Associações | Síndrome de Beckwith-Wiedemann, síndrome de Perlman e hemo-hipertrofia | Síndrome de WAGR, síndrome de Denis-Drash |
VI.1.3 Imuno-histoquímica
Lembrar que, no nefroblastoma, o diagnóstico se fundamenta essencialmente na morfologia à hematoxilina-eosina. Do ponto de vista imuno-histoquímico, podem ser citados:
- WT1:
- Mais importante marcador, não está presente em todos os nefroblastomas.
• Pode estar presente em outras neoplasias.
• Positividade confinada ao núcleo: forte no blastema e epitélio imaturo, e fraca no estroma e epitélio maturo. - Outros: vimentina, CD56, CK e desmina (presentes também na forma blastematosa difusa).
VI.1.4 Biomarcadores
Testes moleculares tais como perda de heterozigozidade nos cromossomos 1p e 16q, ganho 1q e perda 11p15 tem sido intensivamente investigados, sem entretanto resultados definitivos.
Embora os estudos anteriores sejam encorajadores, não existe até o momento um biomarcador definitivo que justifique intervenções terapêuticas estabelecida pelos critérios clássicos. O ganho de 1q e o volume de blastema são no momento os dois fortes candidatos a assumir esse papel futuramente.
VI.2 Outras neoplasias
VI.2.1 Nefroma mesoblástico
- 90% ocorrem antes de 1 ano de idade; muitos são congênitos; predomínio no sexo masculino.
- O tumor pode envolver os vasos do hilo renal.
- Importante a ressecção com margens livres.
- Forma clássica (24% dos casos) – corresponde à fibromatose infantil; média de idade de 3,2 meses.
- Forma celular (66% dos casos) – corresponde ao fibrossarcoma infantil e tem a t(12;15)(p13;q25) com fusão gênica ETV6/NTRK3 e cópias extras dos cromossomos 11, 8 e 17; média de idade de 4,9 meses.
- Forma mista (10% dos casos) – características de ambos os tipos histológicos.
- IIQ: vimentina+; desmina+/-, actina+/-; WT1, EMA, bcl-2, CK, laminina, negativos.
- Cerca de 10% apresentam recorrência, quase sempre no primeiro ano de vida.
VI.2.2 Sarcoma de células claras
- Caracteristicamente, apresenta propensão para metástases ósseas.
• Pico de incidência aos 2 anos de idade. - Não apresentam associações com malformações específicas, ou quadros sindrômicos.
• Sempre unicêntricos. - Espectro histológico amplo simulando outros tumores.
- Cromatina fina com aspecto de núcleo vazado e sequestro de néfrons na interface com o rim – característicos.
- IIQ: vimentina+; NGFR+; CK, EMA, desmina, S100, CD99 negativos. Recentemente tem se demonstrado positividade para ciclina D1 e BCOR
VI.2.3 Tumor rabdoide
- 80% nos primeiros 2 anos de vida; média de idade de 1 ano; predomínio no sexo masculino. Extremamente raro após os 5ª de vida.
• Pode ter associação com tumor na fossa posterior e tumores em tecidos moles.
• Pode ocorrer disseminação cutânea: blueberry muffin rash ou hemangiomatose-símile.
• Prognóstico desfavorável: 80% de óbito em 1 a 2 anos.
• Pode associar-se à síndrome de predisposição rabdoide familiar, associada à inativação de hSFN5/INI1. - Tumor unicêntrico, unilateral, com bordos infiltrativos.
- Núcleo vesicular com nucléolo eosinofílico proeminente e inclusões eosinofílicas citoplasmáticas PAS-positivas.
- IIQ, polifenotípico: vimentina+, EMA+, INI1 negativo; CK/sinaptofisina/desmina/NF/S100, +/-.
- Inativação bialélica do gene supressor de tumor hSNF5/INI1 no cromossomo 22q 11-12, resultante de mutação, deleção, translocação ou perda de todo o cromossomo. Mutações germinativas são associadas à concomitância de tumores renais e do sistema nervoso central (SNC).
- Para ser considerado TR (NWTS-5), deve ter pelo menos dois dos seguintes critérios:
- Histologia consistente com TR.
• IIQ consistente com TR (CK, EMA, actina, NF/sinaptofisina, GFAP).
• INI1 com perda de imunomarcação nuclear.*
*Este anticorpo mostra forte imunomarcação nuclear em virtualmente todos os tipos celulares, exceto o tumor rabdóide, além do carcinoma medular e no sarcoma epitelióide.
VI.2.4 Outros tumores
Outras neoplasias renais primárias malignas – como PNET, sarcoma sinovial, tumor desmoplásico de células pequenas e redondas e os tumores epiteliais (carcinoma de células renais papilífero, carcinoma medular renal, tumores renais com translocação Xp11.2, carcinoma de células renais associado ao neuroblastoma) – e benignas (neoplasias metanéfricas) podem ser infrequentes no rim na faixa etária pediátrica, sendo sugeridas algumas fontes bibliográficas a respeito dessas entidades.
VI.3 Estadiamento
No grupo pediátrico, o sistema de estadiamento TNM não se aplica, devendo-se utilizar o sistema de estadiamento proposto pelo NWTS/COG modificado (SIOP, 2016). O sarcoma de células claras, o nefroma mesoblástico congênito e o tumor rabdoide, juntamente com o nefroblastoma e suas variantes, compreendem 95% de todos os tumores renais primários da infância. Essas neoplasias devem obedecer ao mesmo protocolo macroscópico e de estadiamento que o do TW (Quadro 2). Atualmente, apenas os carcinomas de células renais na infância são estadiados de acordo com o sistema TNM da mesma forma que os pacientes adultos.
Quadro 2 Estadiamento dos tumores renais na infância (exceto os carcinomas)
| Estádio I*: tumor limitado ao rim e completamente ressecado (margens livres) |
| A. O tumor limitado ao rim. |
| B. O tumor está presente na gordura peri-renal mas é revestido por pseudocápsula fibrosa, se fora do contorno normal do rim; a cápsula renal ou pseudocápsula pode estar infiltrada pelo tumor, mas não está transposta; tumor completamente ressecado (margens de ressecção livres)
. C. O tumor pode protruir para a pelve e para o ureter (mas não infiltra sua parede). |
| C. A veia renal e os vasos do seio renal não estão envolvidos. |
| D. O envolvimento de vasos intra-renais pode estar presente. |
| * Nota 1: Biópsia aspirativa por agulha fina ou percutânea (“tru-cut)” não alteram o estadiamento do tumor, mas o diâmetro da agulha deve ser mencionado pelo patologista |
| * Nota 2: Obrigatória a representação de LN regionais (hilo renal, para-aórticos e inguinais) que devem estar livres de neoplasia. |
| Se não receber os LN, o patologista deve incluir todo o tecido adiposo do hilo na tentativa de encontrá-los |
| Estádio II: o tumor estende-se além do rim, mas completamente ressecado (margens livres) e LNs negativos |
| A. Tumor viável está presente na gordura peri-renal e não é recoberto por pseudo-cápsula, mas é completamente ressecado (margens livres). |
| B. Tumor viável infiltra os tecidos moles do seio renal |
| C. Tumor viável presente em vasos do seio renal ou do tecido peri-renal, mas é completamente ressecado. |
| D. O tumor se estende a veia renal ou veia cava inferior, mas é completamente ressecado, não tendo sido transeccionado e não está aderido a parede da veia na margem de ressecção. |
| E. Tumor viável infiltra a parede da pelve renal ou do ureter.
F. Tumor viável infiltra a veia cava ou órgãos adjacentes exceto a glândula adrenal, mas é completamente ressecado |
| Estádio III: tumor residual não hematogênico confinado ao abdome |
| A. Tumor viável presente na margem de ressecção. Tumor não viável ou alterações induzidas pela Qt presentes na margem de ressecção não são considerados estádio III. |
| B. Linfonodo abdominal ou pélvico envolvido, por tumor viável ou não viável. |
| C. Ruptura pré-operatória ou intra-operatória (independentemente de outro critério para estadiamento), se confirmado por exame microscópico (tumor viável na superfície do espécime na área de rotura, identificada pelo cirurgião/tumor removido em mais de uma peça) |
| D. O tumor penetra através da superfície peritoneal |
| E. Trombo tumoral viável presente na margem de ressecção da veia renal, ureter ou veia cava inferior (sempre discutir com cirurgião).
F. Trombo tumoral viável ou não viável aderido a parede da veia cava inferior é retirado em pedaços pelo cirurgião.
G. Biópsia em cunha (“céu aberto”) realizada antes da cirurgia ou Qt pré-operatória. |
| F. Implantes de tumor (viável ou não viável) em qualquer local no abdômen. |
| G. Tumor (viável ou não viável) penetrou através da superfície peritoneal. |
| Estádio IV: metástases hematogênicas (pulmão, fígado, osso, SNC, etc.) |
| A. LN fora da região abdominopélvica.
B. Metástases hematogênicas. |
| Estádio V: envolvimento renal bilateral ao diagnóstico |
| A. Cada lado deve ser estadiado separadamente segundo os critérios citados acima. |
| * Nota: Seio renal: compartimento anatômico que contém a pelve, com tecido adiposo, vasos e nervos. O córtex renal no nível do seio renal é desprovido de cápsula. A presença de nervos é o melhor critério para a identificação do seio renal, pois nunca estão presentes dentro do tumor |
VII.Bibliografia
Ahmed HU, Arya M, Levitt G et al. Part I: Primary malignant non-Wilms’renal tumors in children. Lancet Oncol 2007; 8:730-7.
Ahmed HU, Arya M, Levitt G et al. Part II: Treatment of primary malignant nonWilms´renal tumors in children. Lancet Oncol 2007; 8:842-8.
Antic TA et al. Evolving concepts of cystic renal lesions – the controversy over cystic nephroma and mixed epithelial and stromal tumor of the kidney. Pathol Case Reviews 2006; 11:173-7.
Argani P, Faria PA, Epstein JL et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol 2000; 24:1087-96.
Argani P, Ladany M. Recent advances in pediatric renal neoplasia. Adv Anat Pathol 2003; 10:243-60.
Argani P, Perlman EJ, Breslow NE et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 2000; 24:4-18.
Argani P. Metanephric neoplasms: The hyperdifferentiated, benign end of wilms tumor spectrum? Clin Lab Med 2005; 25:379-92.
Balarezo FS, Joshi VV. Clear cell sarcoma of the pediatric kidney: detailed description and analysis of variant histologic patterns of a tumor with many faces. Adv Anat Pathol 2001; 8:98-108.
Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblatematosis, and the pathogenesis of Wilms’ tumor. Pediatr Pathol 1990; 10:1-36
Beckwith JB, Palmir NF. Histopathology and prognosis of Wilms’ tumour: results of the National Wilms’ Tumour Study Cancer 1978; 41:1937-48.
Bhatnagar S. Management of Wilms’ Tumor: NWTS vs SIOP. J Indian Assoc Pediatr Surg 2009; 14(1):6-14.
Bonsib SM, Pei Y. The non-neoplastic kidney in tumor nephrectomy specimens – what can it show and what is important? Adv Anat Pathol 2010; 17:235-50.
Breslow NE, Beckwith JB, Perlman EJ et al. Age distribution, birth weight, nephrogenic rests and heterogeneity in the pathogenesis of Wilms’ tumor. Ped Blood Cancer 2006; 47:260-7.
D’Angio GJ. The National Wilms Tumor Study: a 40 years perspective. Lifetime Data Anal 2007; 13:463-70.
Eble JN et al. Pathology and genetics of tumours of the urinary system and male genital organs. World Health Organization classification of tumours. Lyon: IARC, 2004.
Faria P, Beckwith JB, Mishra K et al. Focal versus diffuse anaplasia in Wilms tumor – new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol 1996; 20:909-20.
Green DM. Controversies in the management of Wilms tumor: immediate nephrectomy or delayed nephrectomy? Eur J Cancer 2007; 43:2453-6.
Grundy PE, Breslow NE, Li S, Perlman E et al. Loss of heterozigozity for chromosomes 1p and 16q is an adverse prognnostic factor in favorable-histology Wilms Tumor: a report of the National Wilms Tumor Study Group. J Clin Oncol 2005; 29:7312-21.
Herrera GA et al. Renal epithelial tumors 2006. Ancillary diagnostic techniques play a fundamental role in the characterization of a subset of these tumors. Pathol Case Reviews 2006; 11:181-16
Hill A, Amin MB, Bowen J, Dome JS, Grundy PE, Qualman SJ et al.; Members of the Cancer Committee, College of American Pathologists. Protocol for the examination of specimens from pediatric patients with wilms tumors. CAP, 2012. Disponível em: https://www.cap.org/apps/docs/committees/cancer/cancer_protocols/2012/Wilms_12protocol.pdf; acessado em: 13 de fevereiro de 2013.
Hoot AC, Russo P, Judkins AR et al. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 2004; 28:1485-91.
Medeiros LJ, Palmedo G, Krigman HR et al. Oncocytoid renal cell carcinoma after neuroblastoma: a report of four cases of a distinct clinicopathologic entity. Am J Surg Pathol 1999; 23:772-80.
Murphy WM, Grignon DJ, Perlman EJ. Kidney tumors in children. In: Murphy WM, Grignon DJ, Perlman EJ (eds.). Tumors of the kidney, bladder, and related urinary structures. 4. Series, fascicle 1. AFIP atlas of tumor pathology, 2004.
Parham DM, Roloson GJ, Feely M et al. Primary malignant Neuroepithelial Tumors os the kidney: a clinicopathologic analysis of 146 adult and pediatric cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 2001; 25:133-46.
Perlman EJ. Pediatric renal tumors: practical updates for the pathologist. Ped Dev Pathol 2005; 8:320-38.
Protocol for the examination of resection specimens from patients with Wilms and other pediatric renal tumors. College of American Pathologists (CAP); version 4.0.0.0, February 2019.
Qualman SJ, Bowen J, Amin MB, Srigley JR, Grundy PE, Perlman EJ; Members of the Cancer Committee, College of American Pathologists. Protocol for the examination os specimens from patientes with Wilms Tumor (Nephroblastoma) or other renal tumors of chlidhood. Arch Pathol Lab Med 2003; 127:128-89.
Schuetz AN, Yin-Goen Q, Amin MB, Moreno CS, Cohen C, Hornsby CD et al. Molecular classification of renal tumors by gene expression profiling. J Mol Diagn. 2005;7:206-18.
Sebire NJ, Vujanic GM. Paediatric renal tumors: recent developments, new entities and pathological features. Histopathology 2009; 54:516-28.
SIOP/SOBOPE Protocolo clínico para tumores renais – SIOP-RTSG-GBTR 2016. Beatriz de Camargo – bdecamar@terra.com.br
Vujanic GM, Sandstedt B, Harms D, Kelsey A, Leuschner I, de Kraker J; SIOP Nephroblastoma Scientific Committee. Revised International Society of Pediatric Oncology (Siop) working classification of renal tumors of childhood. Med Pediatr Oncol 2002; 38:79-82.
Vujanic GM, Charles AK. Renal Tumors of childhood: un update. Pathology 2008; 40:217-27.
Vujanic G, Sandstedt B. The pathology of Wilms’ tumor (nephroblastoma): the International Society of Paediatric Oncology approach. J Clin Pathol 2010; 63:102-9.
Vujanic G, Gessler M, Ooms AH et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nature Reviews Urology 2018;15:693-701.
Vujanic GM, D’Hooghe E, Popoov SD et al. The effect of preoperative chemotherapy on histological subtyping and staging of Wilms tumors: The United Kingdom Children’s Cancer Study Group (UKCCSG) Wilms tumor trial 3 (UKW3) experience. Ped Blood & Cancer 2019; ;66:e27549.
Weeks DA, Beckwith JB, Mierau GW et al. Rhabdoid tumor of the kidney: a report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol 1989; 13:439-58.
Yan BC, Mackinnon AC, Al-Ahmadie HA et al. Recent developments in the pathology of renal tumors – morphology and molecular characteristics of selected entities. Arch Pathol Lab Med 2009; 133:1026-32
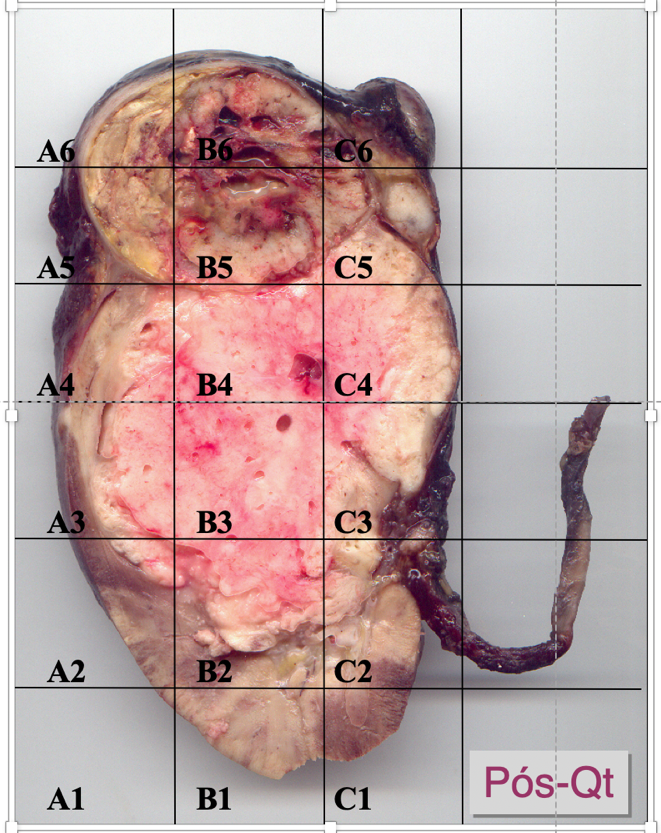
Fig.1 – Mapeamento para representação histológica designada em um espécime de tumor de Wilms
Voltar para a página inicial do manual
